A mixtape for drug discovery
DESY scientists at CFEL have developed a new method that enables automated and fast screening of promising drug candidates. This novel technique, called mix-and-diffuse serial synchrotron crystallography, can image the interaction of potential drug targets with drug candidates or other molecules. The concept has the potential to take structure and fragment based drug design to a new level, as the researchers write in the current issue of the Journal of the International Union of Crystallography (IUCrJ).
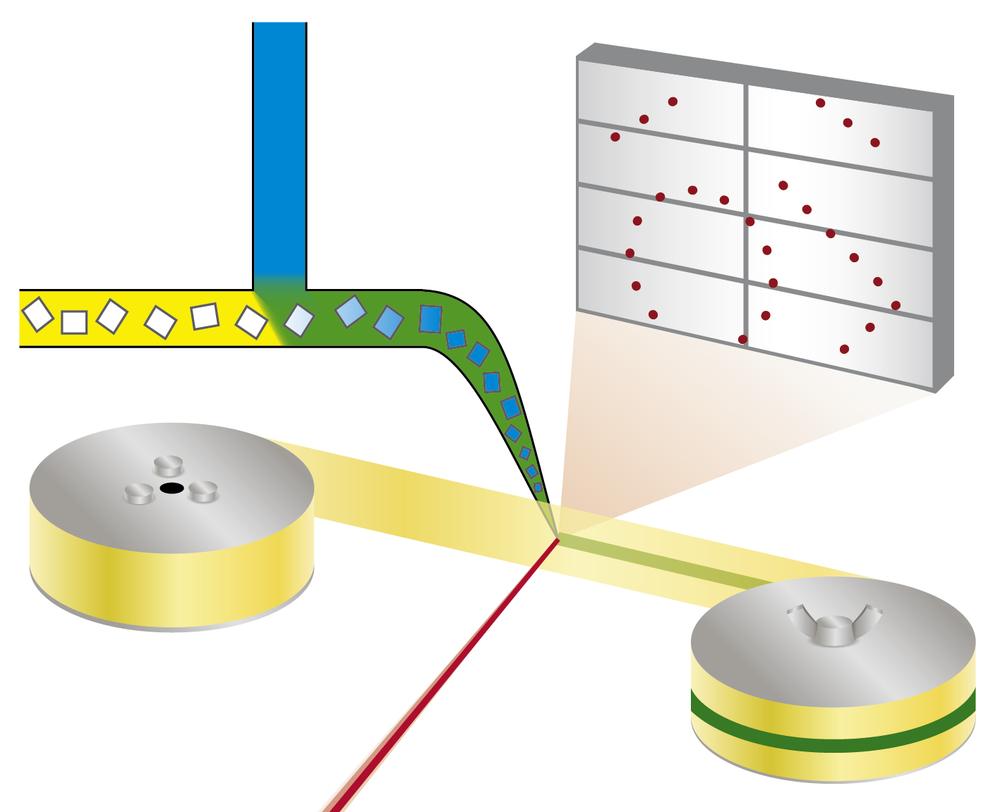
Principle of the mix-and-diffuse serial synchrotron crystallography: protein crystals are mixed with a solution of a drug candidate and X-rayed on a tape running through the X-ray beam. Credit: Beyerlein et al., IUCrJ
Many proteins in the body are potential drug targets. Pharmaceutical molecules with the correct shape can bind to these proteins and activate or deactivate their function. For instance, to combat certain forms of leukaemia, the cancer drug Imatinib inhibits an overactive variant of the enzyme tyrosine-kinase, a protein responsible for activating many other proteins. Imatinib blocks the active site of this tyrosine-kinase. To accomplish this, the drug molecule must precisely fit into the active site like a key into a lock. Based on the knowledge of the spatial structure of the enzyme target, Imatinib was tailor-made for this purpose.
“This strategy is called structure-based drug design and is nowadays used as a standard method in pharmaceutical drug development,” explains first author Kenneth Beyerlein from CFEL. “However, in reality targeting proteins is much more complex than fitting a key into a lock. Therefore, many potential pharmaceutical molecules or fragments of such molecules have to be tested, which usually is a lengthy and complicated procedure.” Furthermore, biologists and pharmacologists alike are interested in the precise workings of natural agents that bind to proteins, to better understand the machinery of life.
The system developed by the team around Beyerlein and his CFEL colleague Dominik Oberthür, among them scientists from the Hamburg Centre for Ultrafast Imaging (CUI), offers a new way of pursuing this goal: It mixes microcrystalline proteins with specific molecules called ligands that can be drug candidates or natural agents just before probing the crystals with X-rays to reveal the detailed spatial structure of the resulting protein-ligand complex or the absence of such a complex if a potential ligand doesn’t bind to the protein.

The enzyme lysozyme (brown) with the inhibiting sugar chitotriose (blue) bound to it. The investigation settled a controversy over the preferred binding site of the sugar molecule. Credit: DESY, Dominik Oberthür
To analyze the spatial structure of a protein, scientists often use X-ray crystallography. For this technique, a crystal has to be grown from the protein first. Researchers then take X-ray snapshots from all sides of the crystal that has to be cooled to ultra-low temperatures to reduce damage from the intense radiation. The X-rays produce a characteristic diffraction pattern from which the inner structure of the crystal and hence the protein's spatial structure can be calculated. To investigate a protein with a ligand, a new crystal has to be grown from a protein and ligand solution or the crystal has to be soaked with the ligand. Even with the use of robotics to automate all steps of this process, the need to mount individual crystals for every new dataset has become the rate limiting step in screening large compound libraries.
The new technique follows a different approach. “We're using microcrystals which has two advantages: They are usually much easier to produce than large crystals, and they are small enough so that a potential drug in a solution can diffuse through the crystal and bind to all protein molecules within in a few milliseconds,” explains Oberthür. The system developed by the team of Oberthür and Beyerlein dispenses a stream of microcrystals in a carrier liquid on a thin tape. Like a conveyor belt, the tape carries the crystals through the X-ray beam, that is chopped into brief flashes by a rotating blind. Instead of rotating a large crystal in the X-ray beam, many microcrystals in random orientation are thus subjected to X-rays in a serial manner and the diffraction patterns from each shot are later combined to form a complete dataset, following the concept of serial crystallography that was first developed at free-electron X-ray lasers (XFELs).
Through a second valve in the system, a solution of a drug candidate or natural ligand is added. The point where the two liquids mix can be adjusted to create a defined delay before investigating the structure. This set-up does not require cryo-cooling of crystals, therefore the protein-drug interaction can be observed at physiological temperatures, or any other desired temperature. This way, even the binding dynamics can be investigated. “We can diffuse chemicals into the protein crystals on the fly and watch the binding happen,” explains Oberthür. “You don't have to find new growth conditions for each inhibitor and you don’t need to exchange the crystals manually, the whole process can be automated.”
The team tested the new system at DESY's high-brilliance X-ray source PETRA III with the well-known protein lysozyme and a sugar molecule, chitotriose, that inhibits the enzyme. The lysozyme microcrystals used here were only about six to eight micrometers in diameter. The set-up at measuring station P11 revealed the spatial structure of the mixed-in inhibitor bound to lysozyme in detail. And even though the structure of lysozyme was the first enzyme structure revealed by X-ray crystallography 50 years ago, the new method could still reveal new details on the binding mode of chitotriose to lysozyme, settling a controversy over the preferred binding site of the sugar molecule.
While the proof of principle still required some time, routine and further advances in detector and X-ray technology will considerably speed up the procedure. Also, using the whole X-ray beam spectrum from the synchrotron light source instead of just a single “colour” from it, can push the exposure time for individual diffraction images down to 100 picoseconds, or 0.1 billionths of a second. Just 50 of these images are sufficient to determine the structure, as was recently shown.
“We're developing ways to solve the structure of bound proteins for high throughput drug discovery,” explained Beyerlein. As synchrotron light sources are more accessible than X-ray lasers, the researchers imagine to use this method for routinely screening through libraries of potential inhibitors and drug fragments. “To do this automatically and much faster than with conventional approaches would be a big step forward in structure-based drug design,” says Beyerlein.
Scientists from California State University, the Norwegian University of Science and Technology, Uppsala University, European XFEL, CFEL, the Hamburg Centre for Ultrafast Imaging, the University of Oxford, SLAC National Accelerator Laboratory, Fachhochschule Lübeck, the University of Hamburg and DESY were involved in the work.